La investigación médico-farmacéutica es un pilar fundamental de la salud pública, un proceso meticuloso y multifacético que nos permite descubrir, desarrollar y evaluar nuevos tratamientos. Comprender sus diferentes tipos y fases es crucial para apreciar el rigor científico que respalda cada medicamento que llega a nuestras manos y, en última instancia, para salvaguardar la seguridad y eficacia de las terapias disponibles.
La investigación médico-farmacéutica se clasifica en fases y diseños de estudio específicos.
- La investigación se estructura en fases preclínica (laboratorio y animales) y clínica (humanos).
- La fase clínica abarca las Fases I (seguridad), II (eficacia preliminar), III (eficacia a gran escala) y IV (vigilancia post-comercialización).
- Los estudios se clasifican también por diseño: experimentales (con intervención, como los ensayos clínicos aleatorizados) u observacionales (sin intervención, solo observación).
- La farmacovigilancia en Fase IV es vital para monitorear la seguridad y los efectos a largo plazo de los fármacos.
- Agencias reguladoras como la AEMPS y la EMA supervisan rigurosamente todo el proceso de desarrollo de medicamentos.
- Las tendencias actuales incluyen el uso de Real-World Evidence (RWE), la medicina personalizada y el desarrollo de terapias génicas y celulares.
El viaje de un fármaco: desde la idea hasta el paciente
El desarrollo de un nuevo medicamento es, en mi experiencia, un viaje extraordinariamente complejo y riguroso, un verdadero ciclo de vida que se extiende desde la identificación inicial de una molécula prometedora hasta su uso seguro y eficaz en pacientes. Este proceso estructurado es vital para garantizar que cada fármaco que llega al mercado haya sido exhaustivamente evaluado en términos de seguridad y eficacia. Las agencias reguladoras, como la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) en España y la Agencia Europea de Medicamentos (EMA) a nivel europeo, juegan un papel indispensable, supervisando cada etapa con un escrutinio implacable para proteger la salud pública.
Investigación preclínica: los cimientos de la seguridad y eficacia
La fase preclínica representa el punto de partida de toda investigación médico-farmacéutica. Es la etapa donde se construyen los cimientos, donde mi equipo y yo nos enfocamos en evaluar la seguridad y la actividad biológica de un compuesto antes de considerar siquiera la posibilidad de probarlo en seres humanos. Aquí es donde se determina si un candidato a fármaco tiene el potencial de ser tanto efectivo como seguro.
Estudios in vitro: ¿Qué revelan las pruebas en el laboratorio?
Los estudios *in vitro* son las primeras pruebas de laboratorio, realizadas en un entorno controlado fuera de un organismo vivo. Pienso en ellos como las "pruebas de concepto" iniciales. Se llevan a cabo en tubos de ensayo, placas de cultivo o modelos celulares, y su función principal es proporcionar una evaluación inicial de la actividad biológica del compuesto y su perfil de seguridad básico. Nos permiten identificar rápidamente si una molécula interactúa con los objetivos biológicos deseados y si presenta alguna toxicidad evidente a nivel celular.
Estudios in vivo: Evaluando seguridad y eficacia en modelos animales
Una vez que un compuesto muestra promesa *in vitro*, el siguiente paso son los estudios *in vivo*, que se realizan en modelos animales. Estos estudios son cruciales para entender cómo el fármaco se comporta en un organismo vivo complejo. Nuestro objetivo aquí es evaluar la seguridad (toxicidad) y la eficacia preliminar del compuesto, observando sus efectos en sistemas biológicos completos antes de dar el salto a los ensayos en humanos. Es una etapa donde la ética y el rigor científico son primordiales.
Farmacocinética y farmacodinámica: Entendiendo la interacción fármaco-cuerpo
Dentro de la fase preclínica, dedicamos una atención significativa a la farmacocinética y la farmacodinámica. La farmacocinética nos dice "lo que el cuerpo le hace al fármaco": cómo se absorbe, se distribuye, se metaboliza y se excreta (ADME). Por otro lado, la farmacodinámica nos revela "lo que el fármaco le hace al cuerpo": sus mecanismos de acción y efectos biológicos. Entender estas dos facetas es fundamental en la fase preclínica, ya que nos proporciona una visión profunda de la interacción fármaco-cuerpo, permitiéndonos predecir su comportamiento en humanos.
¿Cuándo un compuesto está listo para dar el salto a los humanos?
Un compuesto solo está listo para avanzar a los ensayos clínicos en humanos cuando los datos de la fase preclínica son consistentemente positivos y convincentes. Esto significa que hemos obtenido pruebas robustas de seguridad y una clara indicación de actividad biológica en los modelos animales. La rigurosidad de estos datos es innegociable; cualquier señal de toxicidad inaceptable o falta de eficacia nos obliga a reconsiderar o descartar el compuesto, priorizando siempre la seguridad de los futuros pacientes.
Ensayos clínicos: la prueba definitiva en seres humanos
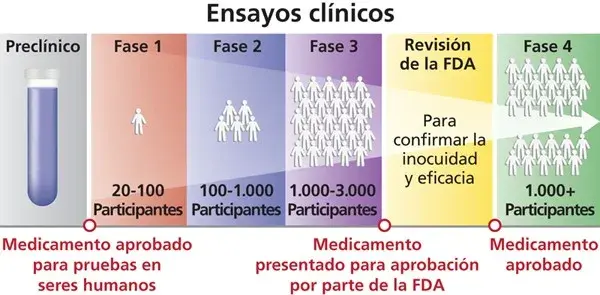
Si un compuesto supera con éxito la fase preclínica, entonces y solo entonces, podemos introducirlo en la fase clínica, donde los fármacos se prueban en seres humanos. Esta etapa es la prueba definitiva y se divide meticulosamente en varias fases, diseñadas para evaluar progresivamente la seguridad y la eficacia del tratamiento en un entorno controlado y ético.
Fase I: El primer contacto con humanos para probar la seguridad
La Fase I de los ensayos clínicos marca el primer contacto de un fármaco con humanos. En esta etapa, el medicamento se administra a un pequeño grupo de voluntarios sanos (generalmente entre 20 y 80 personas). El objetivo principal es evaluar la seguridad del compuesto, determinar un rango de dosificación seguro y comenzar a identificar los efectos secundarios más comunes. Mi enfoque aquí es asegurar que el fármaco sea bien tolerado y que no cause daños inesperados.
Fase II: ¿Realmente funciona? Primeros indicios de eficacia en pacientes
Una vez que la seguridad inicial se ha establecido en Fase I, pasamos a la Fase II. Aquí, el fármaco se administra a un grupo más grande de pacientes (típicamente entre 100 y 300 personas) que padecen la enfermedad o condición para la que se está desarrollando el medicamento. El objetivo primordial de esta fase es obtener datos preliminares sobre la eficacia del tratamiento, al mismo tiempo que continuamos evaluando su seguridad y perfil de efectos secundarios en una población más representativa.
Fase III: La prueba de fuego a gran escala para confirmar beneficios y riesgos
La Fase III es, sin duda, la prueba de fuego. Implica estudios a gran escala, con la participación de entre 1.000 y 3.000 pacientes, a menudo en múltiples centros de investigación a nivel internacional. Sus objetivos son ambiciosos: confirmar la eficacia del fármaco en una población diversa, monitorear de cerca los efectos secundarios a largo plazo o menos comunes, comparar el nuevo tratamiento con los tratamientos estándar existentes y recopilar toda la información necesaria para garantizar su uso seguro. Si los resultados de esta fase son positivos, se puede solicitar la aprobación regulatoria a las agencias pertinentes.
El "estándar de oro": ¿Por qué el Ensayo Clínico Aleatorizado (ECA) es tan importante?
En el ámbito de la investigación clínica, el Ensayo Clínico Aleatorizado (ECA) es considerado el "estándar de oro", y con razón. Se trata de un tipo de estudio experimental donde los participantes son asignados de forma aleatoria a diferentes grupos, por ejemplo, uno que recibe el nuevo tratamiento y otro que recibe un placebo o un tratamiento estándar. Esta asignación aleatoria es crucial porque minimiza el sesgo, asegurando que los grupos sean comparables en todas las características conocidas y desconocidas, lo que permite atribuir con mayor confianza cualquier diferencia en los resultados al efecto del tratamiento en estudio.
Investigación post-aprobación: la vida del fármaco en el mundo real
La aprobación de un medicamento no marca el final de su viaje de investigación; de hecho, es el comienzo de una nueva fase igualmente crítica. La investigación post-aprobación se centra en cómo el fármaco se comporta en la población general, una vez que ha sido comercializado y está disponible para un uso más amplio.
Fase IV: La vigilancia continua tras la comercialización
La Fase IV, también conocida como estudios post-comercialización, es esencial para la seguridad del paciente. En esta etapa, el propósito es recopilar información adicional sobre los riesgos, beneficios y el uso óptimo del fármaco en la población general y a largo plazo. A diferencia de las fases anteriores, que se realizan en poblaciones controladas, la Fase IV nos permite observar el medicamento en un entorno de "mundo real", con una diversidad de pacientes y condiciones que no siempre se replican en los ensayos clínicos.
Farmacovigilancia: Detectando efectos adversos a largo plazo en la población general
Un componente clave de la Fase IV es la farmacovigilancia. Este sistema de vigilancia continua es vital para la salud pública. Su rol principal es la detección, evaluación y prevención de efectos adversos a largo plazo o poco frecuentes que solo se manifiestan una vez que el medicamento es utilizado por una gran cantidad de personas en el mercado. Es un proceso dinámico que permite a las autoridades sanitarias tomar medidas si se identifican nuevos riesgos.
La era del "Real-World Evidence" (RWE): ¿Cómo los datos reales complementan los ensayos?
Actualmente, estamos viviendo la era del "Real-World Evidence" (RWE), que se deriva de los "Real-World Data" (RWD). Estos datos del mundo real provienen de diversas fuentes, como registros electrónicos de salud, bases de datos de reclamaciones de seguros y registros de pacientes. Considero que estos datos son un complemento invaluable a la información obtenida en los ensayos clínicos tradicionales, especialmente en la Fase IV. El RWE nos permite comprender la efectividad y seguridad de los fármacos en la práctica clínica habitual, lo que es crucial para la toma de decisiones regulatorias y de financiación, ofreciendo una perspectiva más completa de cómo un medicamento funciona en la vida diaria de los pacientes.
Tipos de estudios: interviniendo u observando la realidad
Más allá de las fases de desarrollo de un fármaco, la investigación médico-farmacéutica se clasifica también según su diseño metodológico. Esta clasificación nos ayuda a entender cómo se recogen y analizan los datos, y qué tipo de conclusiones podemos extraer de cada estudio.
Estudios experimentales vs. observacionales: ¿Intervenir o solo observar?
La distinción fundamental en el diseño de estudios radica en si el investigador interviene o simplemente observa.
| Estudios Experimentales | Estudios Observacionales |
|---|---|
| El investigador interviene y asigna a los participantes a diferentes grupos (por ejemplo, un grupo recibe el tratamiento y otro un placebo). | El investigador no interviene; solo observa y analiza datos de los pacientes en condiciones reales. |
| Objetivo principal: Evaluar la causalidad entre una intervención (fármaco) y un resultado. | Objetivo principal: Describir la prevalencia, incidencia o asociaciones entre exposiciones y resultados, sin establecer causalidad directa. |
Estudios de cohortes: Siguiendo a grupos a lo largo del tiempo para predecir resultados
Los estudios de cohortes son un tipo de estudio observacional donde se sigue a un grupo de personas (una cohorte) a lo largo del tiempo. Mi equipo y yo los utilizamos para observar quién desarrolla una enfermedad o un resultado específico en relación con una exposición a un fármaco. Son particularmente útiles para estudiar la historia natural de una enfermedad o los efectos a largo plazo de una exposición, ya que permiten establecer una secuencia temporal entre la exposición y el resultado.
Estudios de casos y controles: Mirando hacia atrás para encontrar la causa
En contraste, los estudios de casos y controles son de naturaleza retrospectiva. Aquí, identificamos personas que ya tienen una enfermedad (los "casos") y las comparamos con personas que no la tienen (los "controles"). Luego, investigamos hacia atrás en el tiempo para determinar si hubo diferencias en sus exposiciones pasadas, como el uso de un determinado medicamento. Son eficientes para estudiar enfermedades raras o con largos periodos de latencia.
Estudios transversales: Una fotografía instantánea de la salud de una población
Los estudios transversales nos ofrecen una "fotografía instantánea" de la salud de una población en un único punto en el tiempo. Analizamos datos de una población en un momento específico para determinar la prevalencia de una condición o exposición. Aunque no pueden establecer relaciones de causa y efecto, son excelentes para describir la situación actual de salud o la distribución de características en una población.
El futuro de la farmacéutica: innovaciones y desafíos
El panorama de la investigación médico-farmacéutica está en constante evolución, impulsado por avances tecnológicos y una comprensión más profunda de la biología humana. Veo un futuro emocionante, pero también lleno de desafíos. 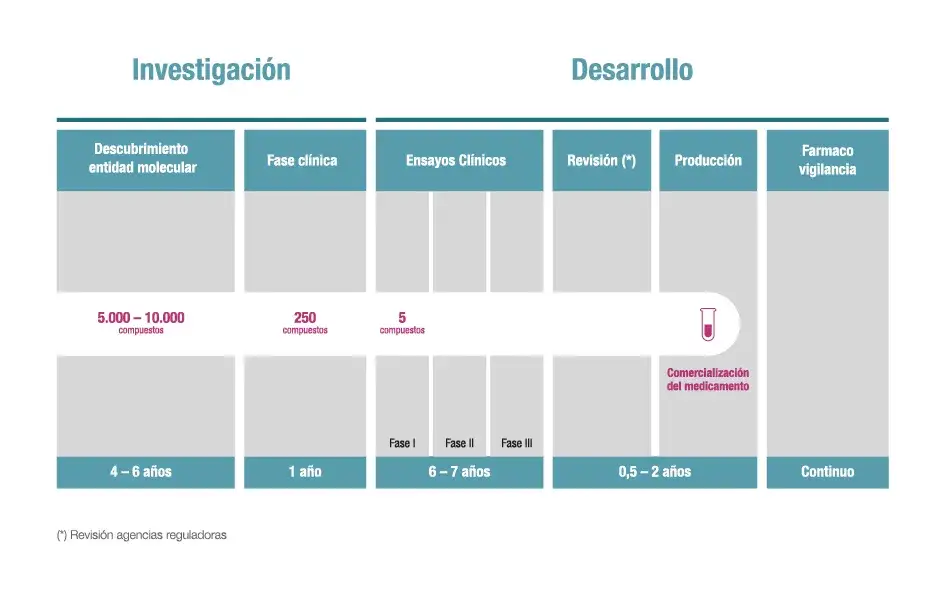
Medicina personalizada: Diseñando fármacos para perfiles genéticos específicos
La medicina personalizada es una de las tendencias más prometedoras y, en mi opinión, transformadoras. Se trata de diseñar fármacos más específicos y eficaces, adaptados a los perfiles genéticos individuales de los pacientes. Al entender las variaciones genéticas que influyen en cómo una persona responde a un tratamiento, podemos optimizar las terapias, minimizando los efectos secundarios y maximizando la eficacia. Es un cambio de paradigma hacia un enfoque mucho más preciso en el tratamiento de enfermedades.
Nuevos horizontes: Terapias génicas, celulares y los desafíos regulatorios que plantean
Los nuevos horizontes en la investigación incluyen las terapias génicas y celulares, que prometen curar enfermedades a nivel fundamental. Estas terapias están impulsando la necesidad de diseños de estudios más complejos y adaptativos, ya que sus mecanismos de acción y perfiles de seguridad son radicalmente diferentes a los de los fármacos tradicionales. Esto, a su vez, presenta desafíos regulatorios significativos, ya que las agencias deben desarrollar marcos para evaluar y aprobar de manera segura y eficiente estas innovaciones revolucionarias. Es un campo donde la ciencia y la regulación deben avanzar de la mano.